Bioinformatics Portfolio
My coursework from BIMM 143. Showcasing genomic analysis techniques, introductory machine learning, and protein folding and structure prediction.
Class 11: AlphaFold
Aadhya Tripathi (PID: A17878439)
- Background
- EBI AlphaFold Database
- Running AlphaFold
- Analysis of Resulting Models
- Predicted Alignment Error for domains
Background
In this hands-on session we will utilize AlphaFold to predict protein structure from sequence (Jumper et al. 2021).
Without the aid of such approaches, it can take years of expensive laboratory work to determine the structure of just one protein. With AlphaFold we can now accurately compute a typical protein structure in as little as ten minutes.
The PDB database (the main repository of experimental structures) only has ~250 thousand structures, as we saw in the last lab. The main protein sequence database has >200 million sequences! Only 0.125% of known sequences have a known structure. This is called the “structure knowledge gap”.
250000 / 200000000
[1] 0.00125
- Structures are much harder to determine than sequences.
- They are expensive (costing $1M on average).
- They take an average of 3-5 years to solve.
EBI AlphaFold Database
The EBI has a database of pre-computed AlphaFold (AF) models called AFDB. This is growing all the time and can be useful to check before running AF ourselves.
Running AlphaFold
We can download and run locally but we need a GPU. Or we can use “cloud” computing to run this on someone else’s computer!
We will use ColabFold https://github.com/sokrypton/ColabFold
We previously found there was no AFDB entry for our HIV sequence.
>HIV-Pr-Dimer
PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF:PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
Here we will use AlphaFold2_mmseqs2.
![]()
Analysis of Resulting Models
Save AlphaFold2 prediction results and import
results_dir <- "HIV_23119/"
pdb_files <- list.files(path=results_dir,
pattern="*.pdb",
full.names = TRUE)
# Print our PDB file names
basename(pdb_files)
[1] "HIV_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb"
[2] "HIV_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb"
[3] "HIV_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb"
[4] "HIV_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb"
[5] "HIV_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb"
Load bio3d and save top 5 ranked models
library(bio3d)
pdbs <- pdbaln(pdb_files, fit=TRUE, exefile="msa")
Reading PDB files:
HIV_23119/HIV_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb
HIV_23119/HIV_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb
HIV_23119/HIV_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb
HIV_23119/HIV_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb
HIV_23119/HIV_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb
.....
Extracting sequences
pdb/seq: 1 name: HIV_23119/HIV_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb
pdb/seq: 2 name: HIV_23119/HIV_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb
pdb/seq: 3 name: HIV_23119/HIV_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb
pdb/seq: 4 name: HIV_23119/HIV_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb
pdb/seq: 5 name: HIV_23119/HIV_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb
pdbs
1 . . . . 50
[Truncated_Name:1]HIV_23119_ PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:2]HIV_23119_ PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:3]HIV_23119_ PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:4]HIV_23119_ PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:5]HIV_23119_ PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
**************************************************
1 . . . . 50
51 . . . . 100
[Truncated_Name:1]HIV_23119_ GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:2]HIV_23119_ GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:3]HIV_23119_ GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:4]HIV_23119_ GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:5]HIV_23119_ GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
**************************************************
51 . . . . 100
101 . . . . 150
[Truncated_Name:1]HIV_23119_ QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:2]HIV_23119_ QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:3]HIV_23119_ QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:4]HIV_23119_ QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:5]HIV_23119_ QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
**************************************************
101 . . . . 150
151 . . . . 198
[Truncated_Name:1]HIV_23119_ GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:2]HIV_23119_ GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:3]HIV_23119_ GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:4]HIV_23119_ GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:5]HIV_23119_ GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
************************************************
151 . . . . 198
Call:
pdbaln(files = pdb_files, fit = TRUE, exefile = "msa")
Class:
pdbs, fasta
Alignment dimensions:
5 sequence rows; 198 position columns (198 non-gap, 0 gap)
+ attr: xyz, resno, b, chain, id, ali, resid, sse, call
Calculate RMSD between all pairs models:
rd <- rmsd(pdbs, fit=T)
Warning in rmsd(pdbs, fit = T): No indices provided, using the 198 non NA positions
range(rd)
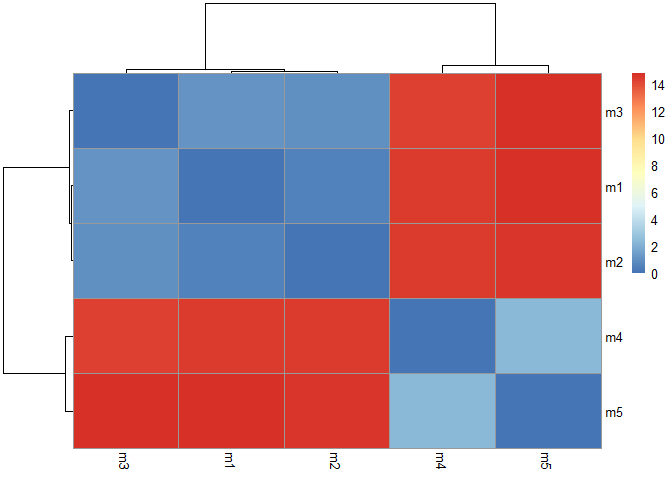
[1] 0.000 14.754
library(pheatmap)
Heatmap of pairs models:
colnames(rd) <- paste0("m",1:5)
rownames(rd) <- paste0("m",1:5)
pheatmap(rd)
# reference PDB structure
pdb <- read.pdb("1hsg")
Note: Accessing on-line PDB file
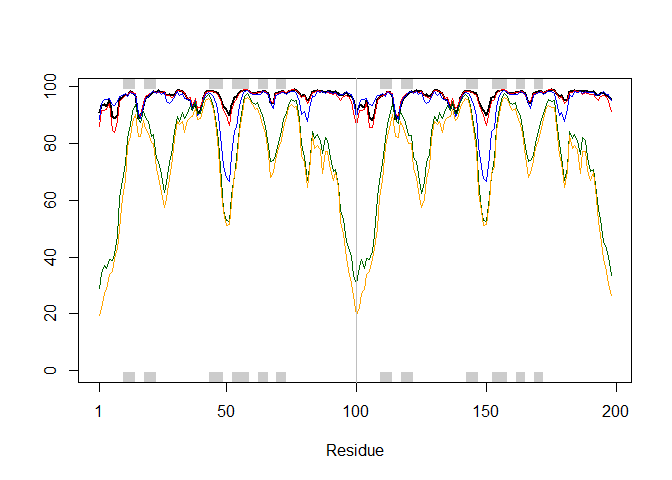
Plot the pLDDT values across all models:
plotb3(pdbs$b[1,], typ="l", lwd=2, sse=pdb)
points(pdbs$b[2,], typ="l", col="red")
points(pdbs$b[3,], typ="l", col="blue")
points(pdbs$b[4,], typ="l", col="darkgreen")
points(pdbs$b[5,], typ="l", col="orange")
abline(v=100, col="gray")
Find most consistent “rigid core” common across all the models:
core <- core.find(pdbs)
core size 197 of 198 vol = 9885.822
core size 196 of 198 vol = 6896.71
core size 195 of 198 vol = 1337.847
core size 194 of 198 vol = 1040.67
core size 193 of 198 vol = 951.857
core size 192 of 198 vol = 899.083
core size 191 of 198 vol = 834.732
core size 190 of 198 vol = 771.338
core size 189 of 198 vol = 733.065
core size 188 of 198 vol = 697.28
core size 187 of 198 vol = 659.742
core size 186 of 198 vol = 625.273
core size 185 of 198 vol = 589.541
core size 184 of 198 vol = 568.253
core size 183 of 198 vol = 545.015
core size 182 of 198 vol = 512.889
core size 181 of 198 vol = 490.723
core size 180 of 198 vol = 470.266
core size 179 of 198 vol = 450.731
core size 178 of 198 vol = 434.735
core size 177 of 198 vol = 420.337
core size 176 of 198 vol = 406.658
core size 175 of 198 vol = 393.334
core size 174 of 198 vol = 382.395
core size 173 of 198 vol = 372.858
core size 172 of 198 vol = 356.994
core size 171 of 198 vol = 346.567
core size 170 of 198 vol = 337.446
core size 169 of 198 vol = 326.659
core size 168 of 198 vol = 314.95
core size 167 of 198 vol = 304.127
core size 166 of 198 vol = 294.552
core size 165 of 198 vol = 285.648
core size 164 of 198 vol = 278.884
core size 163 of 198 vol = 266.765
core size 162 of 198 vol = 258.994
core size 161 of 198 vol = 247.723
core size 160 of 198 vol = 239.84
core size 159 of 198 vol = 234.963
core size 158 of 198 vol = 230.062
core size 157 of 198 vol = 221.985
core size 156 of 198 vol = 215.62
core size 155 of 198 vol = 206.793
core size 154 of 198 vol = 196.984
core size 153 of 198 vol = 188.539
core size 152 of 198 vol = 182.262
core size 151 of 198 vol = 176.954
core size 150 of 198 vol = 170.712
core size 149 of 198 vol = 166.119
core size 148 of 198 vol = 159.796
core size 147 of 198 vol = 153.767
core size 146 of 198 vol = 149.092
core size 145 of 198 vol = 143.657
core size 144 of 198 vol = 137.138
core size 143 of 198 vol = 132.517
core size 142 of 198 vol = 127.231
core size 141 of 198 vol = 121.574
core size 140 of 198 vol = 116.775
core size 139 of 198 vol = 112.57
core size 138 of 198 vol = 108.17
core size 137 of 198 vol = 105.133
core size 136 of 198 vol = 101.249
core size 135 of 198 vol = 97.374
core size 134 of 198 vol = 92.974
core size 133 of 198 vol = 88.184
core size 132 of 198 vol = 84.029
core size 131 of 198 vol = 81.898
core size 130 of 198 vol = 78.019
core size 129 of 198 vol = 75.272
core size 128 of 198 vol = 73.052
core size 127 of 198 vol = 70.695
core size 126 of 198 vol = 68.975
core size 125 of 198 vol = 66.694
core size 124 of 198 vol = 64.394
core size 123 of 198 vol = 62.092
core size 122 of 198 vol = 59.045
core size 121 of 198 vol = 56.629
core size 120 of 198 vol = 54.016
core size 119 of 198 vol = 51.806
core size 118 of 198 vol = 49.652
core size 117 of 198 vol = 48.193
core size 116 of 198 vol = 46.648
core size 115 of 198 vol = 44.752
core size 114 of 198 vol = 43.292
core size 113 of 198 vol = 41.093
core size 112 of 198 vol = 39.147
core size 111 of 198 vol = 36.472
core size 110 of 198 vol = 34.117
core size 109 of 198 vol = 31.47
core size 108 of 198 vol = 29.448
core size 107 of 198 vol = 27.325
core size 106 of 198 vol = 25.822
core size 105 of 198 vol = 24.15
core size 104 of 198 vol = 22.648
core size 103 of 198 vol = 21.069
core size 102 of 198 vol = 19.953
core size 101 of 198 vol = 18.3
core size 100 of 198 vol = 15.723
core size 99 of 198 vol = 14.841
core size 98 of 198 vol = 11.646
core size 97 of 198 vol = 9.434
core size 96 of 198 vol = 7.354
core size 95 of 198 vol = 6.179
core size 94 of 198 vol = 5.666
core size 93 of 198 vol = 4.705
core size 92 of 198 vol = 3.665
core size 91 of 198 vol = 2.77
core size 90 of 198 vol = 2.151
core size 89 of 198 vol = 1.715
core size 88 of 198 vol = 1.15
core size 87 of 198 vol = 0.874
core size 86 of 198 vol = 0.685
core size 85 of 198 vol = 0.528
core size 84 of 198 vol = 0.37
FINISHED: Min vol ( 0.5 ) reached
core.inds <- print(core, vol=0.5)
# 85 positions (cumulative volume <= 0.5 Angstrom^3)
start end length
1 9 49 41
2 52 95 44
xyz <- pdbfit(pdbs, core.inds, outpath="corefit_structures")
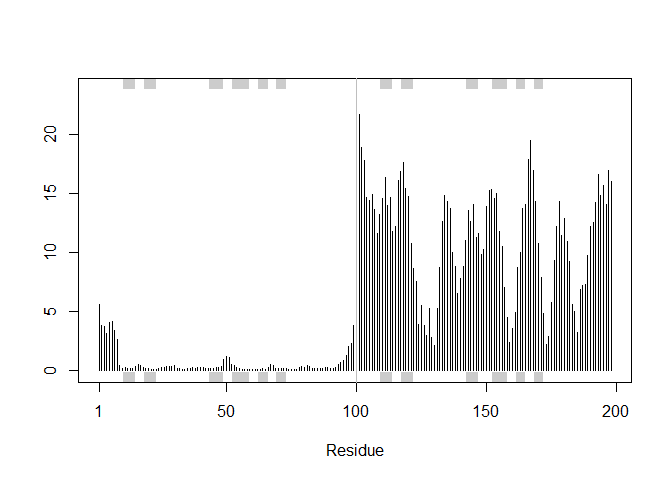
RMSF between positions of the structure:
rf <- rmsf(xyz)
plotb3(rf, sse=pdb)
abline(v=100, col="gray", ylab="RMSF")
Predicted Alignment Error for domains
library(jsonlite)
Access Predicted Aligned Error files:
pae_files <- list.files(path=results_dir,
pattern=".*model.*\\.json",
full.names = TRUE)
PAE files for Model 1 and 5:
pae1 <- read_json(pae_files[1],simplifyVector = TRUE)
pae5 <- read_json(pae_files[5],simplifyVector = TRUE)
attributes(pae1)
$names
[1] "plddt" "max_pae" "pae" "ptm" "iptm"
Plot N by N PAE scores for Model 1:
plot.dmat(pae1$pae,
xlab="Residue Position (i)",
ylab="Residue Position (j)")
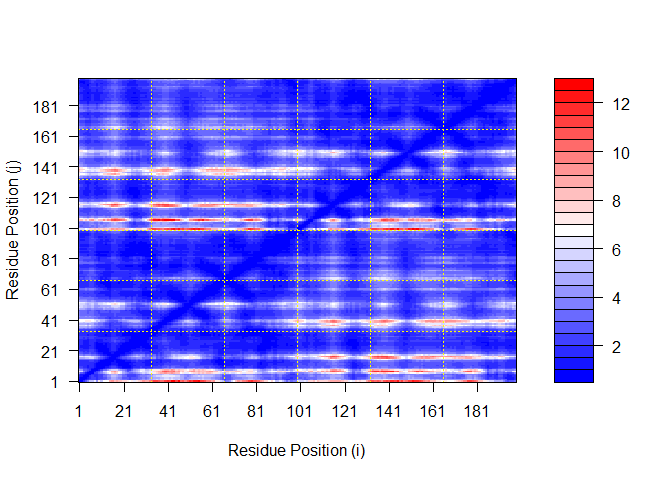
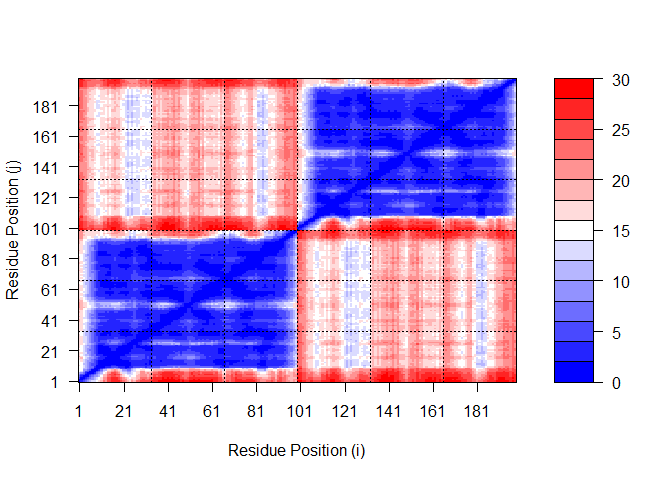
Plot N by N PAE scores for Model 5:
plot.dmat(pae5$pae,
xlab="Residue Position (i)",
ylab="Residue Position (j)",
grid.col = "black",
zlim=c(0,30))
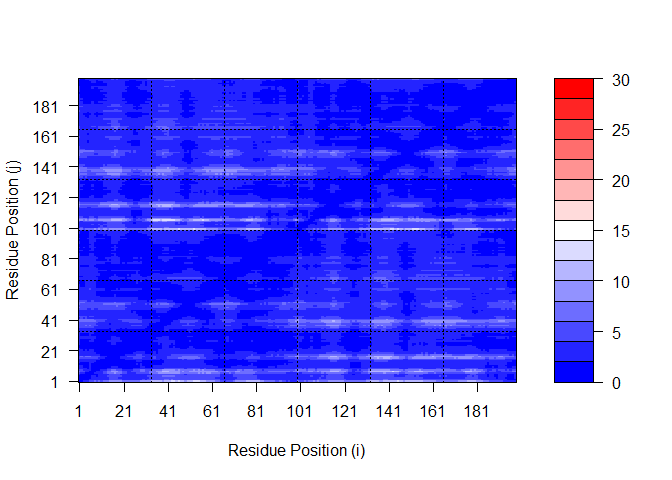
Plot N by N PAE scores for Model 1 again, using the same data range as the plot for model 5:
plot.dmat(pae1$pae,
xlab="Residue Position (i)",
ylab="Residue Position (j)",
grid.col = "black",
zlim=c(0,30))
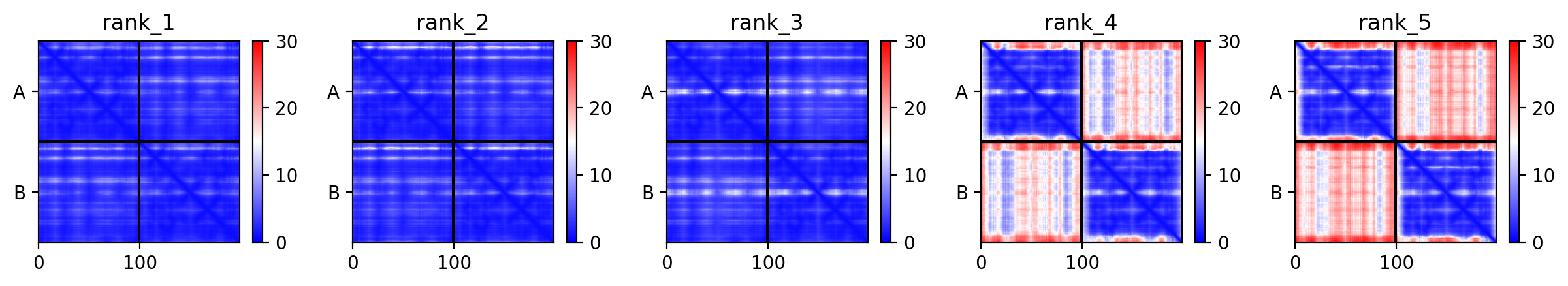 ## Residue
conservation from alignment file
## Residue
conservation from alignment file
aln_file <- list.files(path=results_dir,
pattern=".a3m$",
full.names = TRUE)
aln_file
[1] "HIV_23119/HIV_23119.a3m"
aln <- read.fasta(aln_file[1], to.upper = TRUE)
[1] " ** Duplicated sequence id's: 101 **"
[2] " ** Duplicated sequence id's: 101 **"
Number of sequences in alignment
dim(aln$ali)
[1] 5397 132
Score residue conservation in the alignment
sim <- conserv(aln)
plotb3(sim[1:99], sse=trim.pdb(pdb, chain="A"),
ylab="Conservation Score")
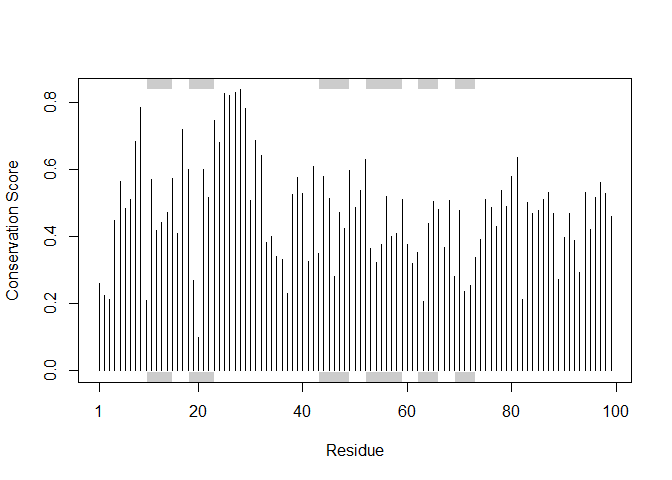
Conserved active sites:
con <- consensus(aln, cutoff = 0.9)
con$seq
[1] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[19] "-" "-" "-" "-" "-" "-" "D" "T" "G" "A" "-" "-" "-" "-" "-" "-" "-" "-"
[37] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[55] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[73] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[91] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[109] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[127] "-" "-" "-" "-" "-" "-"
Save a PDB file with an Occupancy column with conservation scores for visualization.
m1.pdb <- read.pdb(pdb_files[1])
occ <- vec2resno(c(sim[1:99], sim[1:99]), m1.pdb$atom$resno)
write.pdb(m1.pdb, o=occ, file="m1_conserv.pdb")